Methodolgy
We assessed the protein-protein docking benchmark
version 5 for suitable test candidates, i.e. those that had either the receptor
or the ligand as a single chain. In each case we first examined the receptor as
defined in the benchmark, if it was a single chain then it was taken as
suitable for binding site residue prediction against the ligand. If the
receptor was a multi-chain protein then we checked the ligand, if that was a
single chain then it was taken for prediction of the binding site residues, if both
the ligand and receptors were multi-chain then they were rejected. From the 230
complex structures in the benchmark set there were 213 where either the
receptor or ligand were a single chain and thus suitable for prediction.
We then took the single chain from the bound version
of the benchmark structures and using the Ambertools version
20[1] suite of programs
and the Amber FF19SB[2] force-field performed
an energy minimization on the chain. The terminal groups were capped with ACE
and NME residue as were an gaps in the chain, histidines
were protonated according to their protonation state determined by the Reduce
problem. Following 500 steps of steepest descents minimization the resultant
structure was used for an per-residue energy evaluation using the MMPBSA
modules in Ambertools. At the same time the SASA for
each residues was calculated using the FreeSASA
program[3]. Standard
values for the vdw, internal and electrotatic
interactions were pre-calculated for each residue type based on a Me-X-Me
tripeptide and for each residue in the subject protein chain these values were
subtracted from the vdw, internal and electrostatic
interaction energies to give a energy relative to the
standard tri-peptide. Similarly a percentage SASA value was calculated against
the Me-X-Me tri-peptide too.
A conservation score for each residue for the subject
protein chain was calculated using a method similar to ConSurf[4] so for
each residue we now had energy terms and
SASA values relative to the Me-X-Me tripeptide and a conservation score. The
conservation score ranges from one to nine with one being the most variable and
nine being the most conserved. These are the only 3 terms required for the
binding residue prediction.
For the binding residue prediction we rank the
residues in order of their energy deviation from the standard energy from the
most negative to the most positive, the most negative 10% are given a rank of
10, the next 10% 9 and so on until the final 10% are given a rank of 1. The
conservation value is then added to this rank so that the most stable and most
conserved residues have a high score (maximum is 19) and also subtracted so
that the least stable but highly conserved residues have a low score (minimum
is –8). The residues are then reranked by their 2 scores and the equal 4th
highest scoring residues with a SASA of at least 50% are taken along with the
equal 4th lowest scoring residues with a SASA of at least 30%. We
then perform a contact analysis of the residues using HBPlus[5] and pair
up any stable and unstable residues that are within vdw
contact of each other. Then any residue that is within vdw
contact distance of either of the stable/unstable pairing and has a
conservation score of nine is accepted. This gives a minimum of eight potential
binding site residues for each protein chain and can be seen demonstrated in
Figure 1.
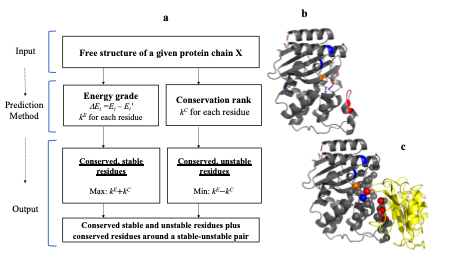
Figure 1 – a) Flowchart for
determining predicted binding residues. The only required input is the PDB
coordinates for the single chain, in this example for PDB code 1JTG chain B. b)
The predicted residues, stable in blue, unstable in red and bridge in orange,
c) predicted residues vs actual binding site ones depicted as spheres, the
ligand is in yellow.
To locate the binding residues themselves we used PyMOL to determine all residues on the subject chain that
had a heavy-atom to heavy-atom distance of £
5.0 Å from the bound version of the complex to the other chains present. These
residues were then determined to be actual binding residues for that chain.
The predicted residues using only the energy and conservation
evaluations were then compared with the actual binding residues from the 3D
complex structure and a residue was determined to be a positive mactch if it was either a binding residue or its immediate neighbour was. This criteria allowed for areas of protein
where there were residue gaps in binding residue numbers such as 1A2K where the
a range of binding residues included 61–63, 65–68 and 70–73 so that a
prediction of residue 69, 70, 71, 73 and 74 would all be taken as positive
predictions.
References
[1] Case DA, Belfon K, Ben-Shalom IY, Brozell SR, Cerutti DS, Cheatham I,
T.E. , et al. AMBER 2020. Amber. 20 ed:
University of California, San Francisco.; 2020.
[2] Tian
C, Kasavajhala K, Belfon KAA, Raguette L, Huang H, Migues AN, et al. ff19SB:
Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum
Mechanics Energy Surfaces in Solution. Journal of Chemical Theory and
Computation. 2020;16:528-52.
[3]
Mitternacht S. FreeSASA: An open source C library for solvent accessible
surface area calculations. F1000Research. 2016:S 189.
[4]
Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016:
an improved methodology to estimate and visualize evolutionary conservation in
macromolecules. Nucleic Acids Res. 2016;44:W344–W50.
[5]
McDonald IK, Thornton JM. Satisfying hydrogen bonding potential in proteins. J
Mol Biol. 1994;238:777-93.